


 Sign Out
Sign Out

PHARMACOLOGY: Mode of Action: Empagliflozin is a reversible, highly potent and selective competitive inhibitor of SGLT2 with an IC50 of 1.3 nM. It has a 5000-fold selectivity over human SGLT1 (IC50 of 6278 nM), responsible for glucose absorption in the gut. Furthermore high selectivity could be shown toward other glucose transporters (GLUTs) responsible for glucose homeostasis in the different tissues.
SGLT-2 is highly expressed in the kidney, whereas expression in other tissues is absent or very low. It is responsible as the predominant transporter for reabsorption of glucose from the glomerular filtrate back into the circulation. In patients with type 2 diabetes mellitus (T2DM) and hyperglycaemia a higher amount of glucose is filtered and reabsorbed.
Empagliflozin improves glycaemic control in patients with T2DM by reducing renal glucose reabsorption. The amount of glucose removed by the kidney through this glucuretic mechanism is dependent upon the blood glucose concentration and GFR. Through inhibition of SGLT-2 in patients with T2DM and hyperglycaemia, excess glucose is excreted in the urine.
In patients with T2DM, urinary glucose excretion increased immediately following the first dose of empagliflozin and is continuous over the 24 hour dosing interval. Increased urinary glucose excretion was maintained at the end of 4-week treatment period, averaging approximately 78 g/day with 25 mg empagliflozin once daily. Increased urinary glucose excretion resulted in an immediate reduction in plasma glucose levels in patients with T2DM.
Empagliflozin improves both fasting and post-prandial plasma glucose levels.
The mechanism of action of empagliflozin is independent of beta cell function and insulin pathway and this contributes to a low risk of hypoglycaemia.
Improvement of surrogate markers of beta cell function including Homeostasis Model Assessment-B (HOMA-β) and proinsulin to insulin ratio were noted. In addition urinary glucose excretion triggers calorie loss, associated with body fat loss and body weight reduction.
The glucosuria observed with empagliflozin is accompanied by mild diuresis which may contribute to sustained and moderate reduction of blood pressure.
Metformin is a biguanide with antihyperglycaemic effects, lowering both basal and post-prandial plasma glucose. It does not stimulate insulin secretion and therefore does not produce hypoglycaemia.
Metformin hydrochloride may act via 3 mechanisms: (1) reduction of hepatic glucose production by inhibiting gluconeogenesis and glycogenolysis; (2) in muscle, by increasing insulin sensitivity, improving peripheral glucose uptake and utilisation; (3) and delay of intestinal glucose absorption.
Metformin hydrochloride stimulates intracellular glycogen synthesis by acting on glycogen synthase.
Metformin hydrochloride increases the transport capacity of all types of membrane glucose transporters (GLUTs) known to date.
In humans, independently of its action on glycaemia, metformin hydrochloride has favourable effects on lipid metabolism. This has been shown at therapeutic doses in controlled, medium-term or long-term clinical studies: metformin hydrochloride reduces total cholesterol, LDL cholesterol and triglyceride levels.
Clinical Trials: A total of 10224 patients with type 2 diabetes were treated in 9 double-blind, placebo or active-controlled clinical studies of at least 24 weeks duration, of which 2947 patients received empagliflozin 10 mg and 3703 received empagliflozin 25 mg as add-on to metformin therapy.
Treatment with empagliflozin in combination with metformin with or without other background (pioglitazone, sulfonylurea, DPP-4 inhibitors, and insulin) led to clinically relevant improvements in HbA1c, fasting plasma glucose, body weight, systolic and diastolic blood pressure. Administration of empagliflozin 25 mg resulted in a higher proportion of patients achieving HbA1c goal of <7% and fewer patients needing glycaemic rescue compared to empagliflozin 10 mg and placebo. There was a clinically meaningful improvement in HbA1c in all subgroups of gender, race, geographic region, time since diagnosis of T2DM and body mass index (BMI). In patients aged 75 years and older, numerically lower reductions in HbA1c were observed with empagliflozin treatment. Higher baseline HbA1c was associated with a greater reduction in HbA1c. Empagliflozin in combination with metformin in drug-naïve patients led to clinically meaningful reductions in HbA1c, FPG, body weight and BP.
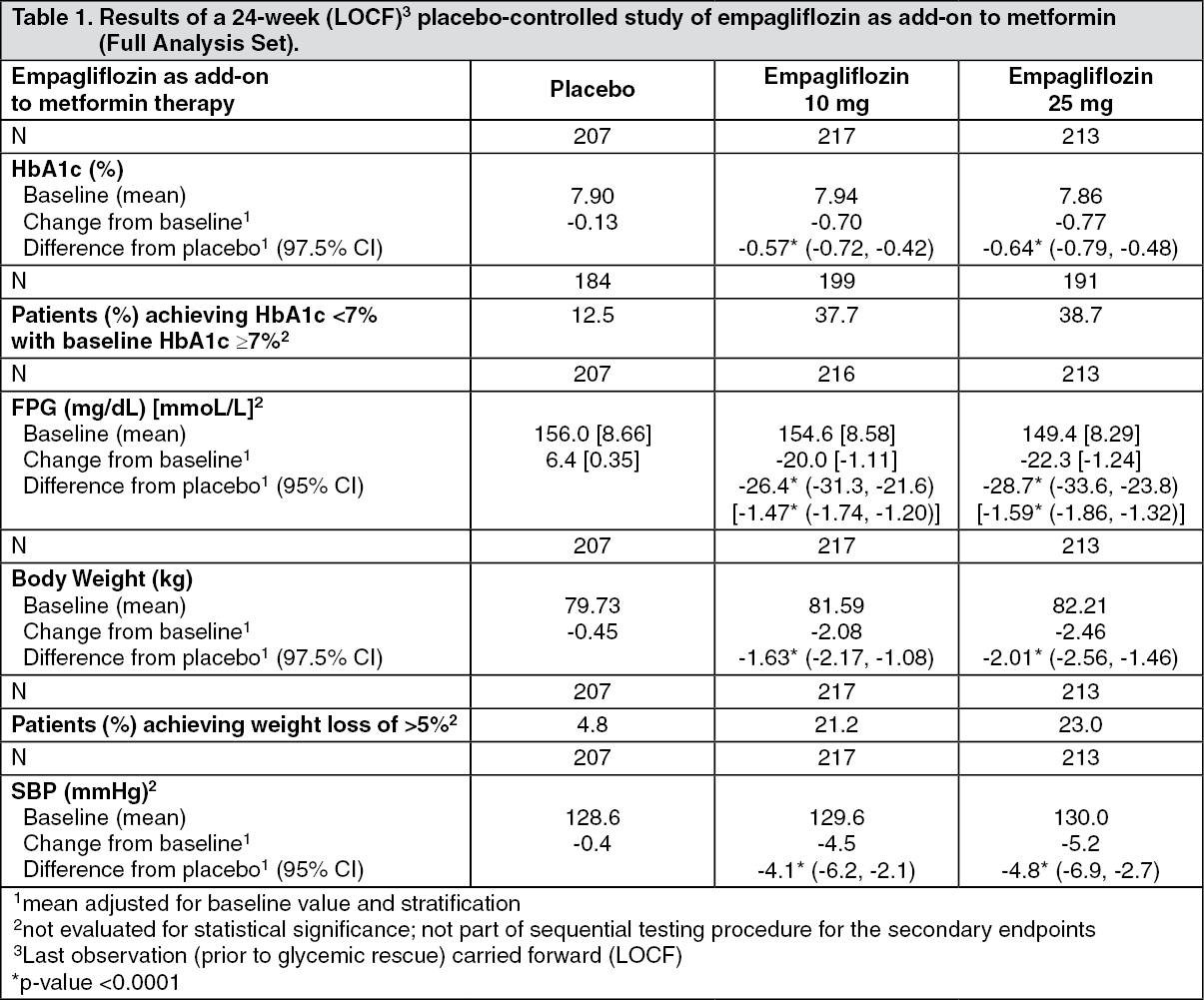
Empagliflozin as add-on to metformin therapy: A double-blind, placebo-controlled study of 24 weeks duration was conducted to evaluate the efficacy and safety of empagliflozin in patients not sufficiently treated with metformin. Treatment with empagliflozin resulted in statistically significant improvements in HbA1c and body weight, and clinically meaningful reductions in FPG and blood pressure compared to placebo (Table 1).
In the double-blind placebo-controlled extension of this study, reductions of HbA1c (change from baseline of -0.62% for empagliflozin 10 mg, -0.74% for empagliflozin 25 mg and -0.01% for placebo), body weight (change from baseline of -2.39 kg for empagliflozin 10 mg, -2.65 kg for empagliflozin 25 mg and -0.46 kg for placebo) and blood pressure (SBP: change from baseline of -5.2 mmHg for empagliflozin 10 mg, -4.5 mmHg for empagliflozin 25 mg and -0.8 mmHg for placebo, DBP: change from baseline of -2.5 mmHg for empagliflozin 10 mg, -1.9 mmHg for empagliflozin 25 mg and -0.5 mmHg for placebo) were sustained up to Week 76. (See Table 1.)
 Click on icon to see table/diagram/image
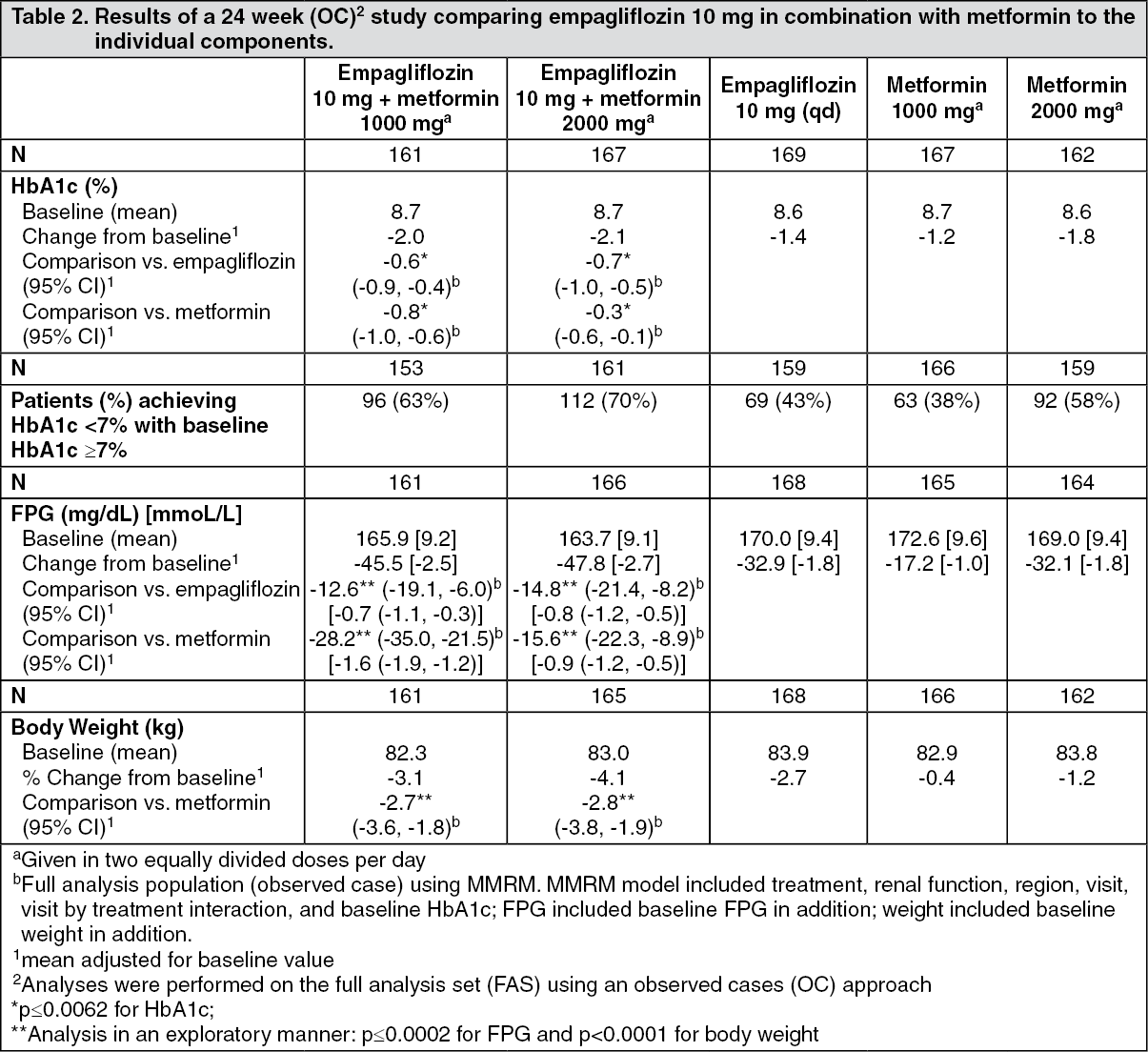
Click on icon to see table/diagram/imageEmpagliflozin and metformin combination therapy in drug-naïve patients: A factorial design study of 24 weeks duration was conducted to evaluate the efficacy and safety of empagliflozin in drug-naïve patients. Treatment with empagliflozin in combination with metformin (5 mg and 500 mg; 5 mg and 1000 mg; 12.5 mg and 500 mg, and 12.5 mg and 1000 mg given twice daily) provided statistically significant improvements in HbA1c and led to significantly greater reductions in FPG and body weight compared to the individual components. A greater proportion of patients with a baseline HbA1c ≥ 7.0% and treated with empagliflozin in combination with metformin achieved a target HbA1c < 7% compared to the individual components (see Tables 2 and 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image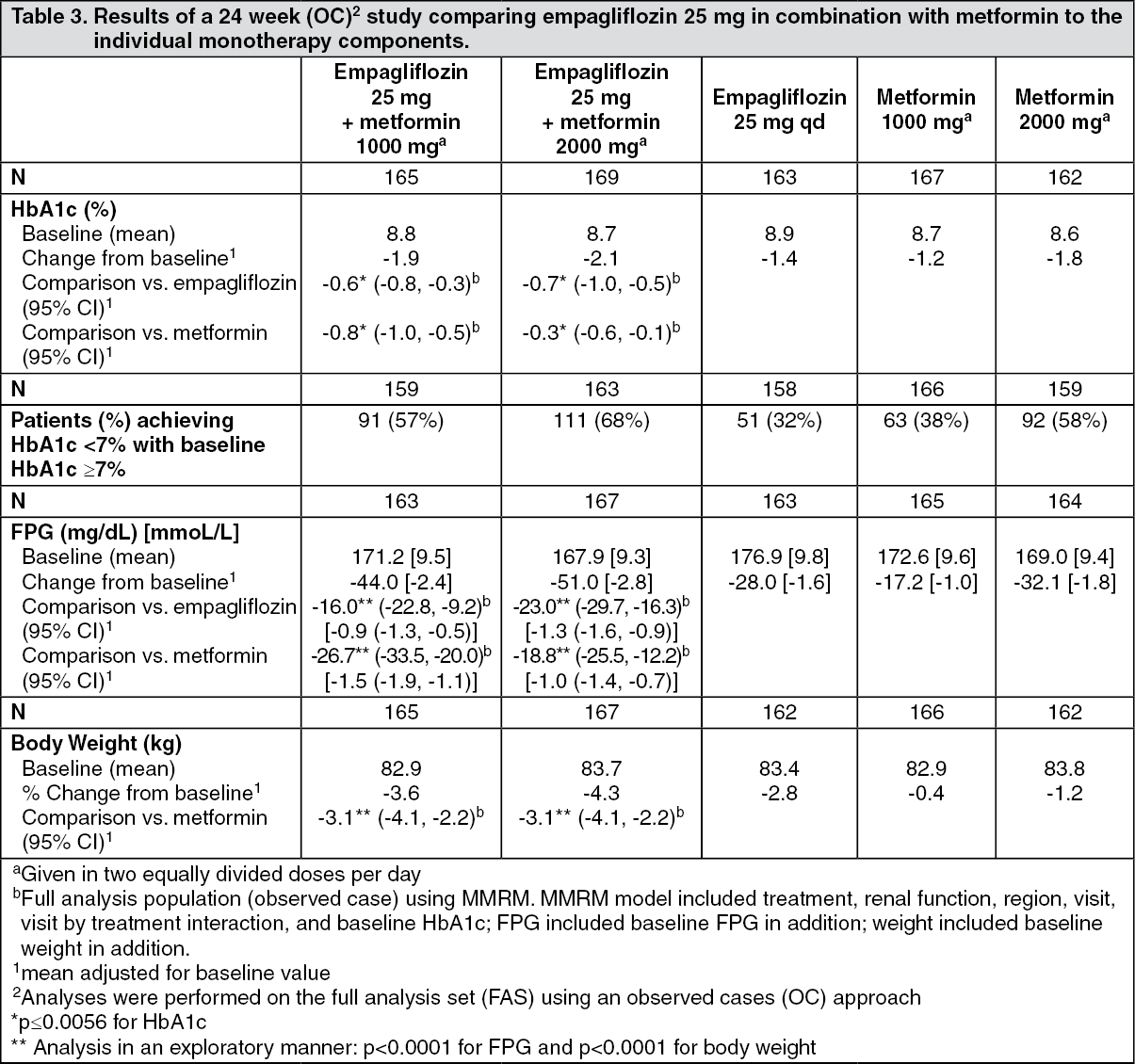 Click on icon to see table/diagram/image
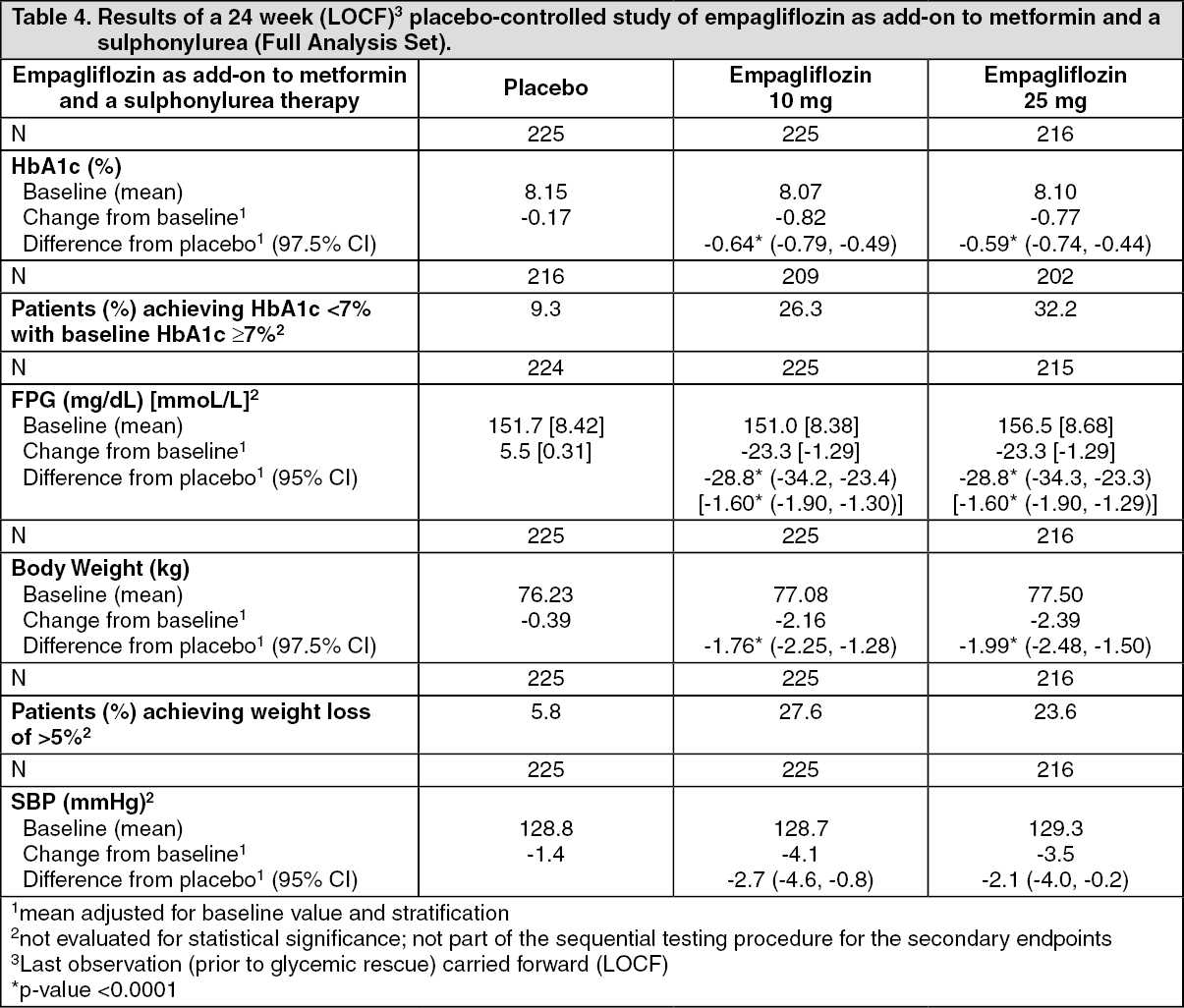
Click on icon to see table/diagram/imageEmpagliflozin as add-on to a combination of metformin and sulphonylurea therapy: A double-blind, placebo-controlled study of 24 weeks duration was conducted to evaluate the efficacy and safety of empagliflozin in patients not sufficiently treated with a combination of metformin and a sulphonylurea. Treatment with empagliflozin resulted in statistically significant improvements in HbA1c and body weight and clinically meaningful reductions in FPG and blood pressure compared to placebo (Table 4).
In the double-blind placebo-controlled extension of this study, reductions of HbA1c (change from baseline of -0.74% for empagliflozin 10 mg, -0.72% for empagliflozin 25 mg and -0.03% for placebo), body weight (change from baseline of -2.44 kg for empagliflozin 10 mg, -2.28 kg for empagliflozin 25 mg and -0.63 kg for placebo) and blood pressure (SBP: change from baseline of -3.8 mmHg for empagliflozin 10 mg, -3.7 mmHg for empagliflozin 25 mg and -1.6 mmHg for placebo, DBP: change from baseline of -2.6 mmHg for empagliflozin 10 mg, -2.3 mmHg for empagliflozin 25 mg and -1.4 mmHg for placebo) were sustained up to Week 76. (See Table 4.)
 Click on icon to see table/diagram/image
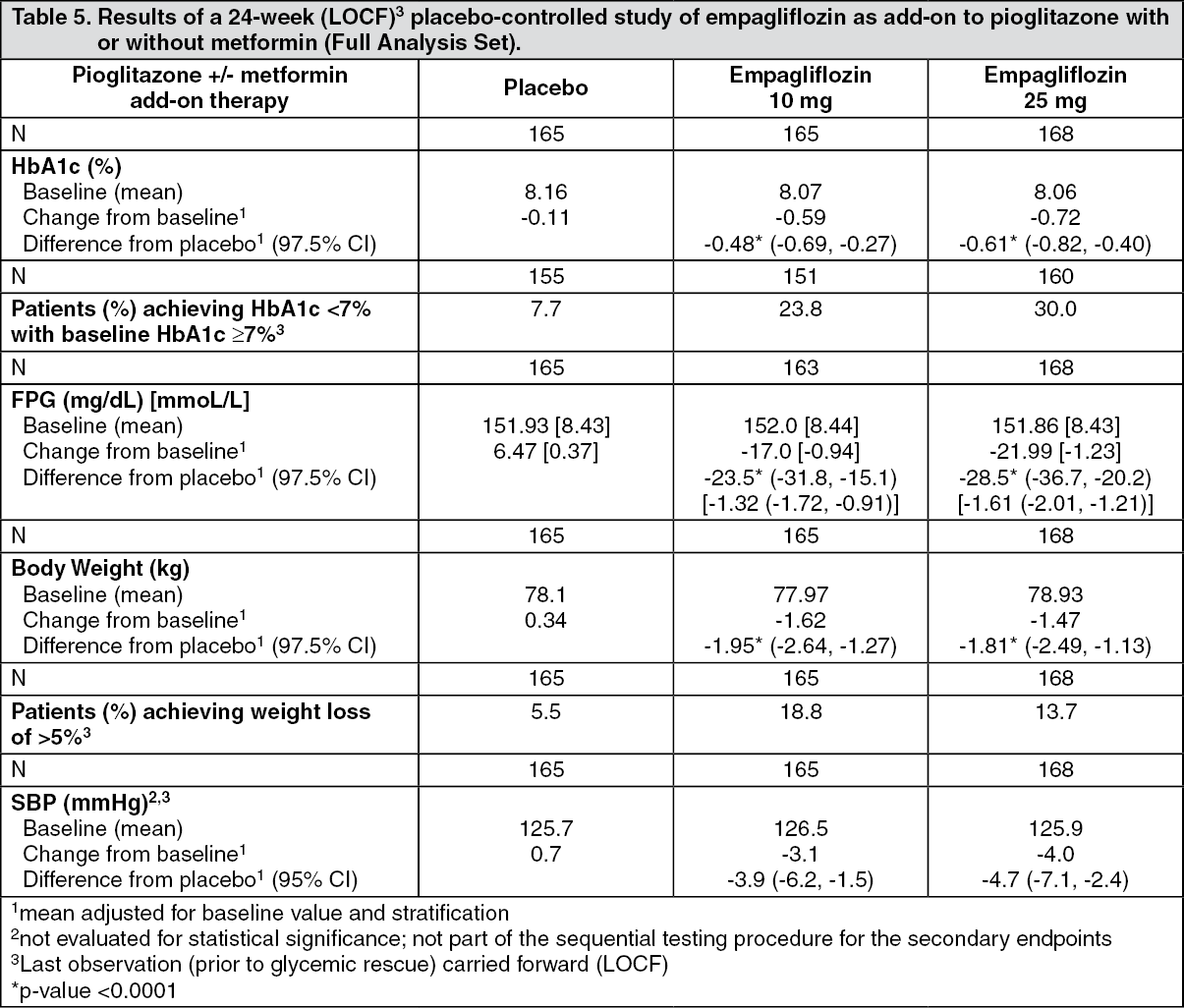
Click on icon to see table/diagram/imageEmpagliflozin as add-on to a combination of pioglitazone therapy (+/- metformin): The efficacy and safety of empagliflozin in combination with pioglitazone, with or without metformin (75.5% of all patients were on metformin background) was evaluated in a double-blind, placebo-controlled study of 24 weeks duration. Empagliflozin in combination with pioglitazone (dose ≥ 30 mg) with or without metformin resulted in statistically significant reductions in HbA1c, fasting plasma glucose, and body weight and clinically meaningful reductions in blood pressure compared to placebo (Table 5).
In the double-blind placebo-controlled extension of this study, reductions of HbA1c (change from baseline of -0.61% for empagliflozin 10 mg, -0.70% for empagliflozin 25 mg and -0.01% for placebo), body weight (change from baseline of -1.47 kg for empagliflozin 10 mg, -1.21 kg for empagliflozin 25 mg and +0.50 kg for placebo) and blood pressure (SBP: change from baseline of -1.7 mmHg for empagliflozin 10 mg, -3.4 mmHg for empagliflozin 25 mg and +0.3 mmHg for placebo, DBP: change from baseline of -1.3 mmHg for empagliflozin 10 mg, -2.0 mmHg for empagliflozin 25 mg and +0.2 mmHg for placebo) were sustained up to Week 76. (See Table 5.)
 Click on icon to see table/diagram/image
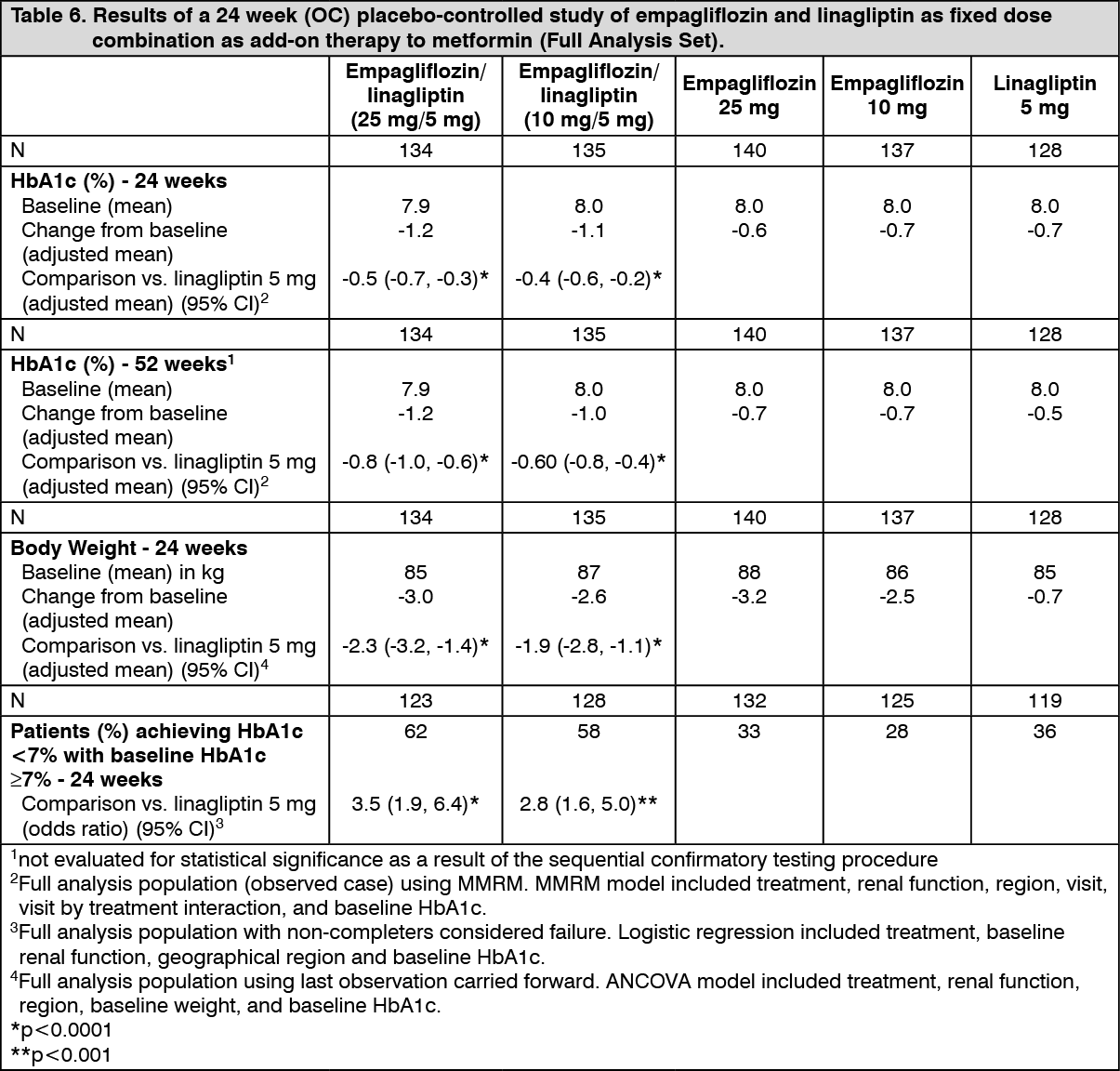
Click on icon to see table/diagram/imageEmpagliflozin and linagliptin as add-on therapy to metformin: In a factorial design study, patients inadequately controlled on metformin, 24-weeks treatment with both doses of empagliflozin 10 mg and 25 mg administered together with linagliptin 5 mg provided statistically significant improvements in HbA1c and FPG compared to linagliptin 5 mg and also compared to empagliflozin 10 or 25 mg. Compared to linagliptin 5 mg, both doses of empagliflozin plus linagliptin 5 mg provided statistically significant reductions in body weight and blood pressure. A greater proportion of patients with a baseline HbA1c ≥ 7.0% and treated with empagliflozin plus linagliptin achieved a target HbA1c of < 7% compared to linagliptin 5 mg (Table 6).
After 24 weeks' treatment with empagliflozin+linagliptin, both systolic and diastolic blood pressures were reduced, -5.6/-3.6 mmHg (p<0.001 versus linagliptin 5 mg for SBP and DBP) for empagliflozin 25 mg+linagliptin 5 mg and -4.1/-2.6 mmHg (p<0.05 versus linagliptin 5 mg for SBP, n.s. for DBP) for empagliflozin 10 mg+linagliptin 5 mg. Clinically meaningful reductions in blood pressure were maintained for 52 weeks, -3.8/-1.6 mmHg (p<0.05 versus linagliptin 5 mg for SBP and DBP) for empagliflozin 25 mg/linagliptin 5 mg and -3.1/-1.6 mmHg (p<0.05 versus linagliptin 5 mg for SBP, n.s. for DBP) for empagliflozin 10 mg/linagliptin 5 mg.
After 24 weeks, rescue therapy was used in 1 (0.7%) patient treated with empagliflozin 25 mg/linagliptin 5 mg and in 3 (2.2%) patients treated with empagliflozin 10 mg/linagliptin 5 mg, compared to 4 (3.1%) patients treated with linagliptin 5 mg and 6 (4.3%) patients treated with empagliflozin 25 mg and 1 (0.7%) patient treated with empagliflozin 10 mg. (See Table 6.)
 Click on icon to see table/diagram/image
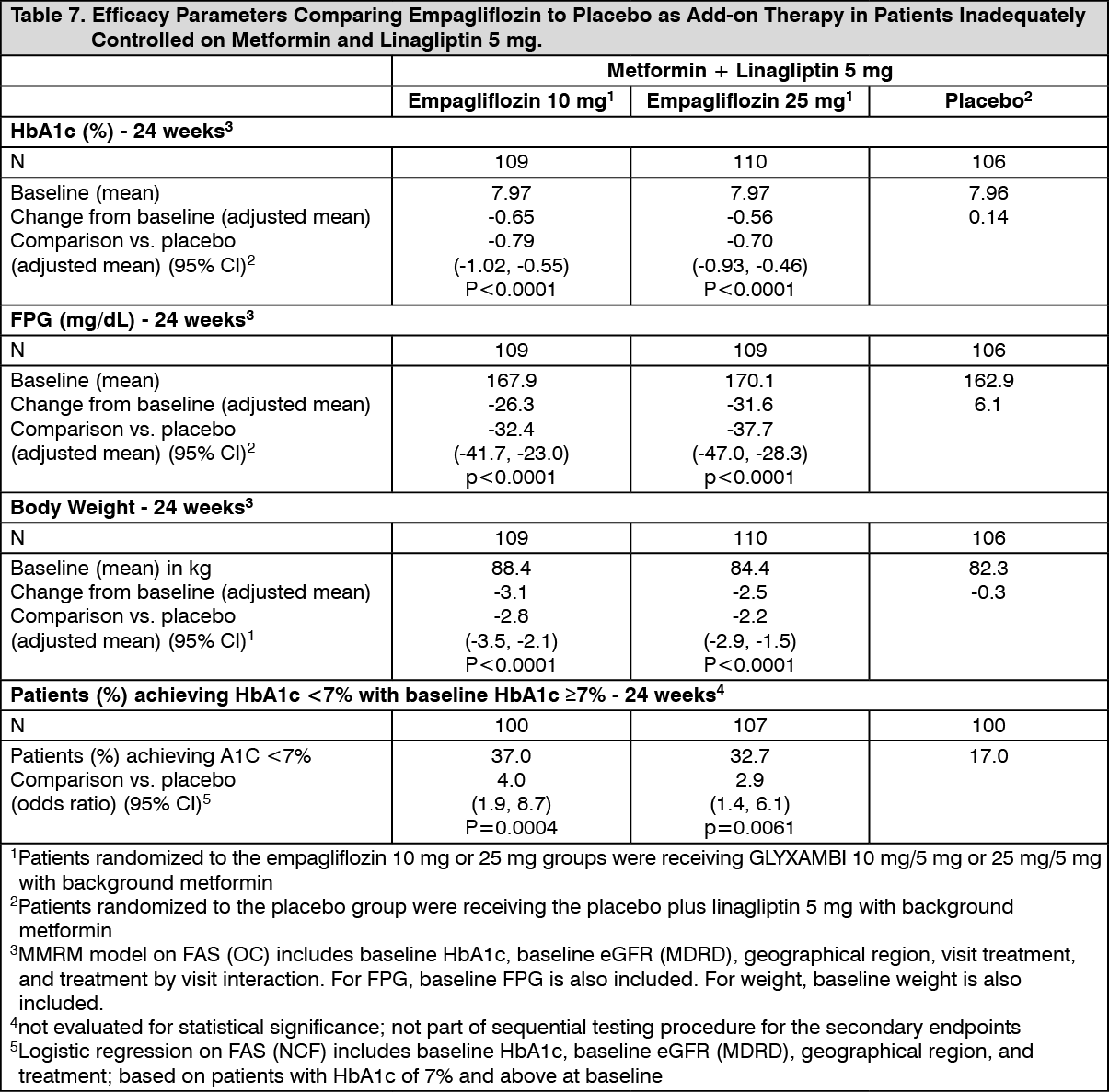
Click on icon to see table/diagram/imageEmpagliflozin in patients inadequately controlled on metformin and linagliptin: In patients inadequately controlled on metformin and linagliptin 5 mg, 24-weeks treatment with both GLYXAMBI 10 mg/5mg and GLYXAMBI 25 mg/5 mg provided statistically significant improvements in HbA1c, FPG and body weight compared to placebo+linagliptin 5 mg. A statistically significantly greater number of patients with a baseline HbA1c ≥7.0% and treated with both doses of empagliflozin achieved a target HbA1c of <7% compared to placebo+linagliptin 5 mg (Table 7). After 24 weeks' treatment with empagliflozin, both systolic and diastolic blood pressures were reduced, -2.6/-1.1 mmHg (n.s. versus placebo for SBP and DBP) for empagliflozin 25 mg+linagliptin 5 mg and -1.3/-0.1 mmHg (n.s. versus placebo for SBP and DBP) for empagliflozin 10 mg+linagliptin 5 mg.
After 24 weeks, rescue therapy was used in 4 (3.6%) patients treated with empagliflozin 25 mg+linagliptin 5 mg and in 2 (1.8%) patients treated with empagliflozin 10 mg+linagliptin 5 mg, compared to 13 (12.0%) patients treated with placebo+linagliptin 5 mg. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn a prespecified subgroup of patients with baseline HbA1c greater or equal than 8.5% the reduction from baseline in HbA1c with empagliflozin 25 mg+linagliptin 5 mg was -1.3% at 24 weeks (p<0.0001 versus placebo+linagliptin 5 mg) and with empagliflozin 10 mg+linagliptin 5 mg -1.3% at 24 weeks (p<0.0001 versus placebo+linagliptin 5 mg).
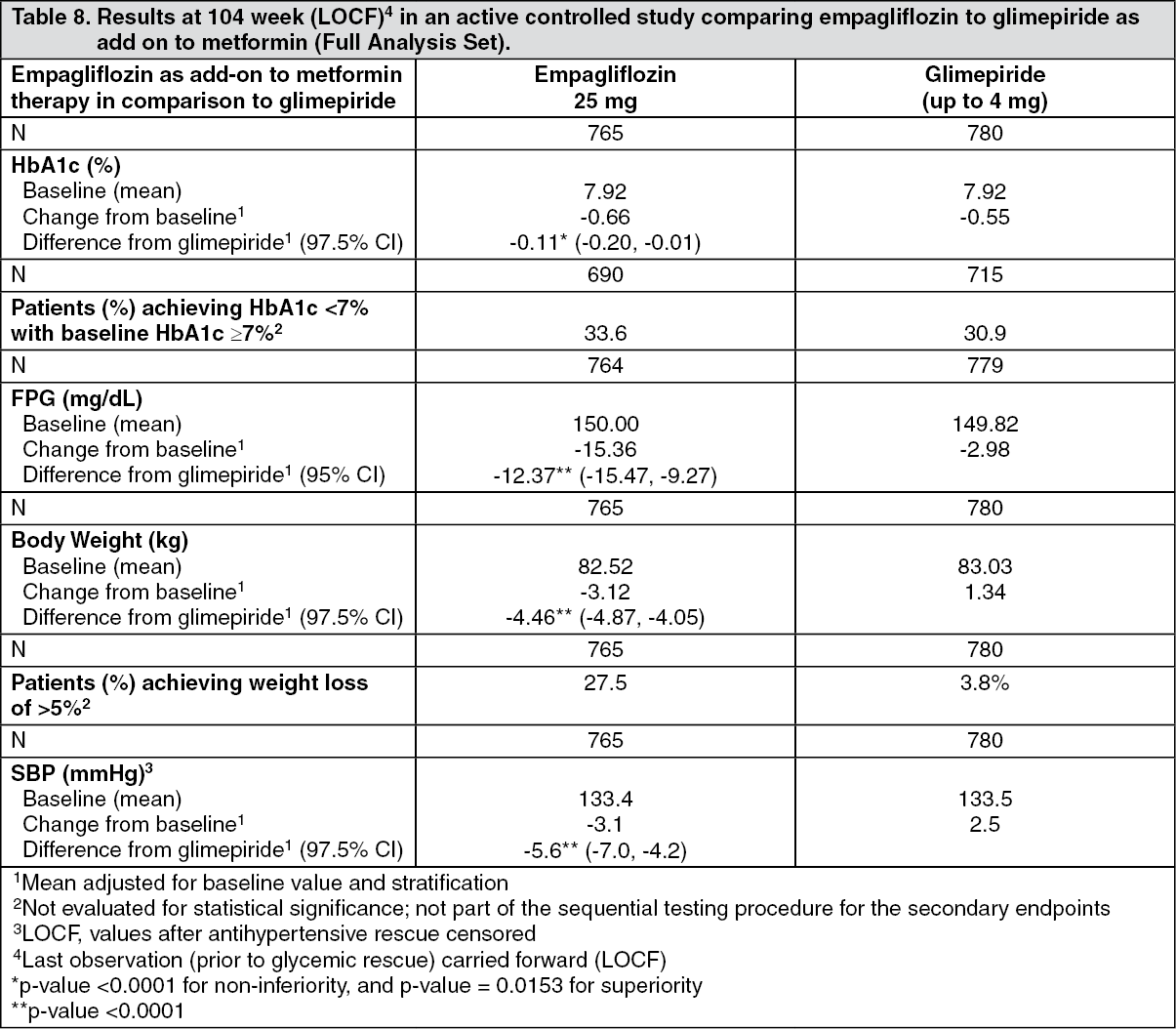
Empagliflozin 2-year data, as add-on to metformin in comparison to glimepiride: In a study comparing the efficacy and safety of empagliflozin 25 mg versus glimepiride (4 mg) in patients with inadequate glycaemic control on metformin alone, treatment with empagliflozin daily resulted in superior reduction in HbA1c, and a clinically meaningful reduction in FPG, compared to glimepiride (Table 8). Empagliflozin daily resulted in a statistically significant reduction in body weight, systolic and diastolic blood pressure (change from baseline in DBP of -1.8 mmHg for empagliflozin and +0.9 mmHg for glimepiride, p < 0.0001).
Treatment with empagliflozin resulted in statistically significantly lower proportion of patients with hypoglycaemic events compared to glimepiride (2.5% for empagliflozin, 24.2% for glimepiride, p < 0.0001). (See Table 8.)
 Click on icon to see table/diagram/image
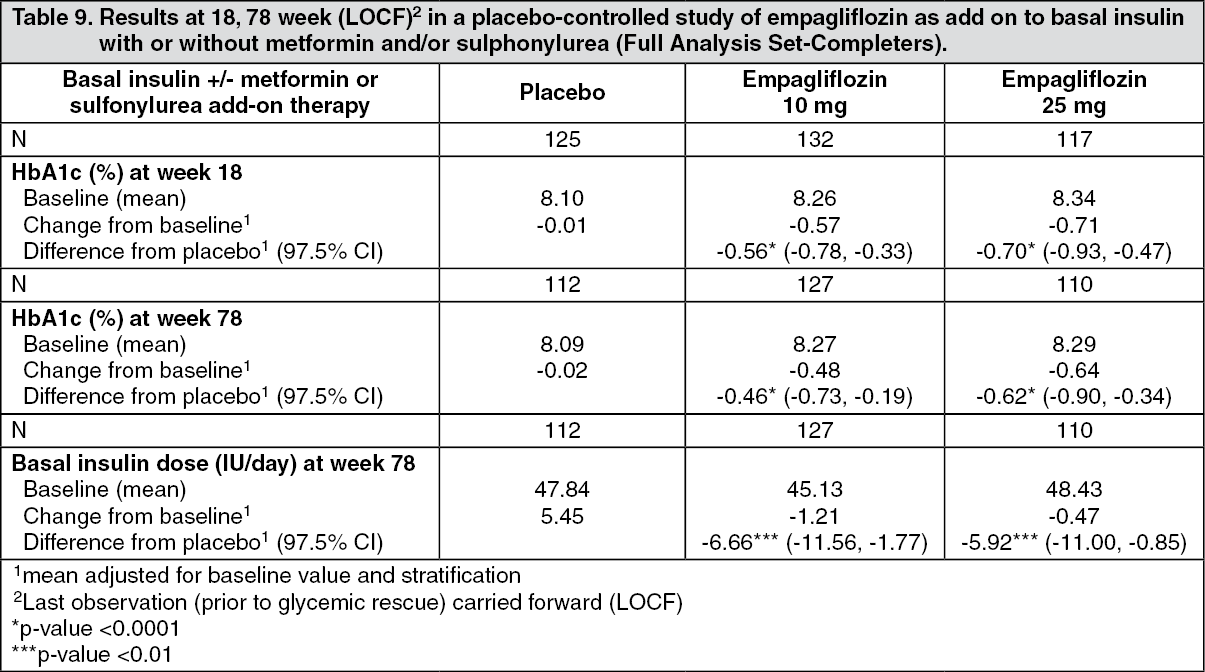
Click on icon to see table/diagram/imageEmpagliflozin as add-on to basal insulin therapy: The efficacy and safety of empagliflozin as add-on to basal insulin with or without concomitant metformin and/or sulfonylurea therapy (79.8% of all patients were on metformin background) was evaluated in a double-blind, placebo-controlled trial of 78 weeks duration. During the initial 18 weeks the insulin dose was to be kept stable, but was adjusted to achieve a FPG < 110 mg/dL in the following 60 weeks.
At week 18, empagliflozin provided statistically significant improvement in HbA1c compared to placebo. A greater proportion of patients with a baseline HbA1c ≥ 7.0% achieved a target HbA1c of < 7% compared to placebo. At 78 weeks, empagliflozin resulted in a statistically significant decrease in HbA1c and insulin sparing compared to placebo (Table 9).
At week 78, empagliflozin resulted in a reduction in FPG (-10.51 mg/dL [-0.58 mmoL/L] for empagliflozin 10 mg, -17.43 mg/dL [0.3 mmoL/L] for empagliflozin 25 mg and -5.48 mg/dL [-0.97 mmoL/L] for placebo), body weight (-2.47 kg for empagliflozin 10 mg, -1.96 kg for empagliflozin 25 mg and +1.16 kg for placebo, p < 0.0001), blood pressure (SBP: -4.1 mmHg for empagliflozin 10 mg, -2.4 mmHg for empagliflozin 25 mg and +0.1 mmHg for placebo, DPB: -2.9 mmHg for empagliflozin 10 mg, -1.5 mmHg for empagliflozin 25 mg and -0.3 mmHg for placebo). (See Table 9.)
 Click on icon to see table/diagram/image
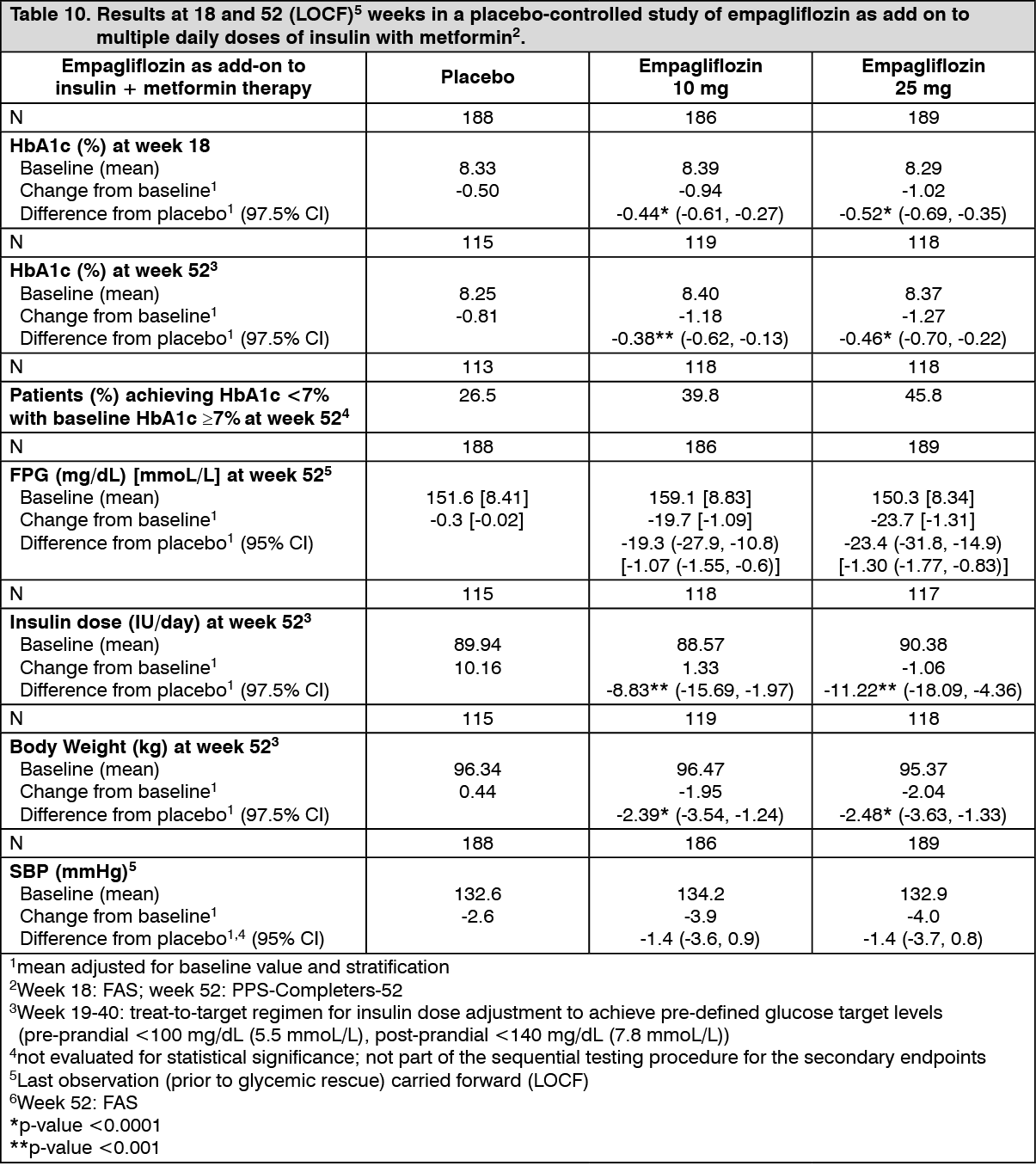
Click on icon to see table/diagram/imageEmpagliflozin as add-on to MDI insulin therapy and metformin: The efficacy and safety of empagliflozin as add-on to multiple daily insulin with or without concomitant metformin therapy (71.0% of all patients were on metformin background) was evaluated in a double-blind, placebo-controlled trial of 52 weeks duration. During the initial 18 weeks and the last 12 weeks, the insulin dose was kept stable, but was adjusted to achieve pre-prandial glucose levels < 100 mg/dL [5.5 mmoL/L], and post-prandial glucose levels < 140 mg/dL [7.8 mmoL/L] between Weeks 19 and 40.
At Week 18, empagliflozin provided statistically significant improvement in HbA1c compared with placebo (Table 10). A greater proportion of patients with a baseline HbA1c ≥ 7.0% (19.5% empagliflozin 10 mg, 31.0% empagliflozin 25 mg) achieved a target HbA1c of < 7% compared with placebo (15.1%).
At Week 52, treatment with empagliflozin resulted in a statistically significant decrease in HbA1c and insulin sparing compared with placebo and a reduction in FPG (change from baseline of -0.3 mg/dL [-0.02 mmoL/L] for placebo, -19.7 mg/dL [-1.09 mmoL/L] for empagliflozin 10 mg, and -23.7 mg/dL [-1.31 mmoL/L] for empagliflozin 25 mg), body weight, and blood pressure (SBP: change from baseline of -2.6 mmHg for placebo, -3.9 mmHg for empagliflozin 10 mg and 4.0 mmHg for empagliflozin 25 mg, DBP: change from baseline of -1.0 mmHg for placebo, -1.4 mmHg for empagliflozin 10 mg and -2.6 mmHg for empagliflozin 25 mg). (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEmpagliflozin twice daily versus once daily as add-on to metformin therapy: The efficacy and safety of empagliflozin twice daily versus once daily (daily dose of 10 mg and 25 mg) as add-on therapy in patients with insufficient glycaemic control on metformin monotherapy was evaluated in a double-blind placebo-controlled study of 16 weeks duration. All treatments with empagliflozin resulted in significant reductions in HbA1c from baseline (total mean 7.8%) after 16 weeks of treatment compared with placebo. Empagliflozin twice daily dose regimens led to comparable reductions in HbA1c versus once daily dose regimens with a treatment difference in HbA1c reductions from baseline to week 16 of -0.02% (95% CI -0.16, 0.13) for empagliflozin 5 mg twice daily vs. 10 mg once daily, and -0.11% (95% CI -0.26, 0.03) for empagliflozin 12.5 mg twice daily vs. 25 mg once daily.
2 hour post-prandial glucose: Treatment with empagliflozin as add-on to metformin or metformin plus sulfonylurea resulted in clinically meaningful improvement of 2-hour post-prandial glucose (meal tolerance test) at 24 weeks (add-on to metformin, placebo (n=57): +5.9 mg/dL, empagliflozin 10 mg (n=52): -46.0 mg/dL, empagliflozin 25 mg (n=58): -44.6 mg/dL; add-on to metformin plus sulphonylurea, placebo (n=35): -2.3 mg/dL, empagliflozin 10 mg (n=44): -35.7 mg/dL, empagliflozin 25 mg (n=46): -36.6 mg/dL).
Patients with baseline HbA1c ≥ 9%: In a pre-specified analysis of subjects with baseline HbA1c ≥ 9.0%, treatment with empagliflozin 10 mg or 25 mg as add-on to metformin resulted in statistically significant reductions in HbA1c at Week 24 (adjusted mean change from baseline of -1.49% for empagliflozin 25 mg, -1.40% for empagliflozin 10 mg, and -0.44% for placebo).
Body weight: In a pre-specified pooled analysis of 4 placebo-controlled studies, treatment with empagliflozin (68% of all patients were on metformin background) resulted in body weight reduction compared to placebo at week 24 (-2.04 kg for empagliflozin 10 mg, -2.26 kg for empagliflozin 25 mg and -0.24 kg for placebo) that was maintained up to week 52 (-1.96 kg for empagliflozin 10 mg, -2.25 kg for empagliflozin 25 mg and -0.16 kg for placebo).
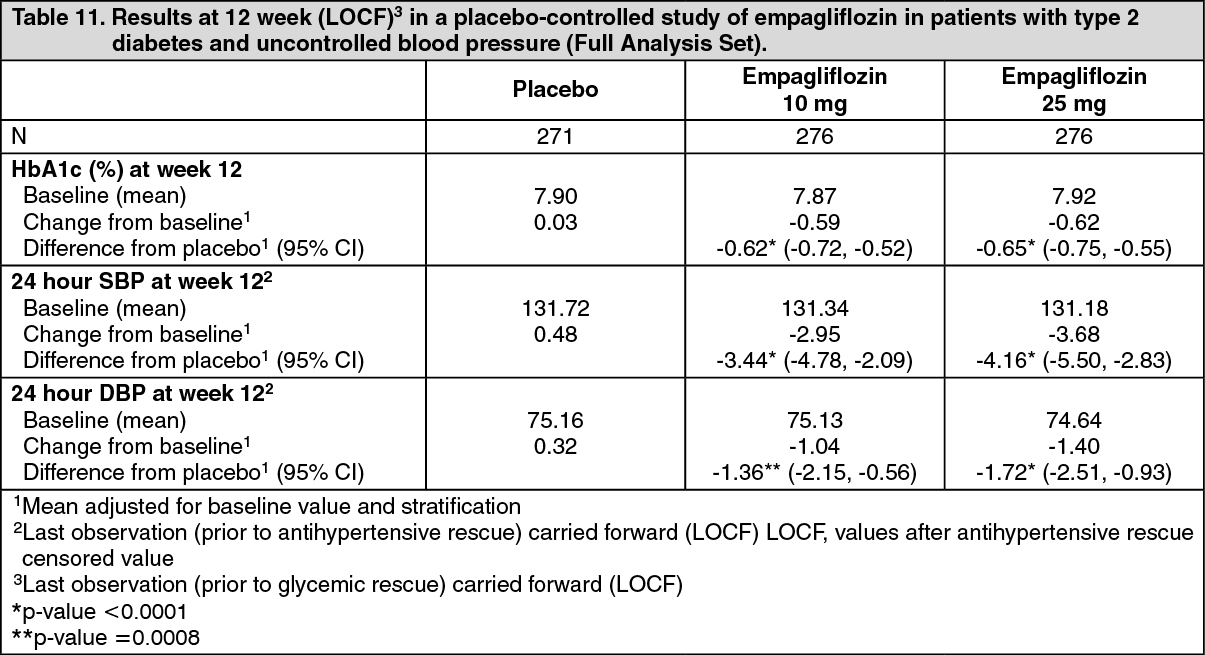
Blood pressure: The efficacy and safety of empagliflozin was evaluated in a double-blind, placebo-controlled study of 12 weeks duration in patients with type 2 diabetes and high blood pressure on different antidiabetic (67.8% treated with metformin with or without other antidiabetic drugs including insulin) and up to 2 antihypertensive therapies (Table 11). Treatment with empagliflozin once daily resulted in statistically significant improvement in HbA1c, 24 hour mean systolic and diastolic blood pressure as determined by ambulatory blood pressure monitoring. Treatment with empagliflozin provided reductions in seated SBP (change from baseline of -0.67 mmHg for placebo, -4.60 mmHg for empagliflozin 10 mg and -5.47 mmHg for empagliflozin 25 mg) and seated DBP (change from baseline of -1.13 mmHg for placebo, -3.06 mmHg for empagliflozin 10 mg and -3.02 mmHg for empagliflozin 25 mg). (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn a pre-specified pooled analysis of 4 placebo-controlled studies, treatment with empagliflozin (68% of all patients were on metformin background) resulted in a reduction in systolic blood pressure (empagliflozin 10 mg -3.9 mmHg, empagliflozin 25 mg -4.3 mmHg) compared with placebo (-0.5 mmHg), and in diastolic blood pressure (empagliflozin 10 mg -1.8 mmHg, empagliflozin 25 mg -2.0 mmHg) compared with placebo (-0.5 mmHg), at week 24, that were maintained up to week 76.
Laboratory parameters: Haematocrit increased: In a pooled safety analysis of all trials with metformin background treatment, mean changes from baseline in haematocrit were 3.6% and 4.0% for empagliflozin 10 mg and 25 mg, respectively, compared to 0% for placebo. In the EMPA-REG Outcome study, haematocrit values returned towards baseline values after a follow-up period of 30 days after treatment stop.
Serum lipids increased: In a pooled safety analysis of all trials with metformin background treatment, mean percent increases from baseline for empagliflozin 10 mg and 25 mg versus placebo, respectively, were total cholesterol 5.0% and 5.2% versus 3.7%; HDL-cholesterol 4.6% and 2.7% versus -0.5%; LDL-cholesterol 9.1% and 8.7% versus 7.8%; triglycerides 5.4% and 10.8% versus 12.1%.
Cardiovascular outcome: The EMPA-REG OUTCOME study is a multi-centre, multi-national, randomized, double-blind, placebo-controlled trial investigating the effect of empagliflozin as adjunct to standard care therapy in reducing cardiovascular events in patients with type 2 diabetes and one or more cardiovascular risk factors, including coronary artery disease, peripheral artery disease, history of myocardial infarction (MI), or history of stroke. The primary endpoint was the time to first event in the composite of CV death, non-fatal MI, or non-fatal stroke (Major Adverse Cardiovascular Events (MACE-3)). Additional pre-specified endpoints addressing clinically relevant outcomes tested in an exploratory manner included CV death, the composite of heart failure requiring hospitalization or CV death, all-cause mortality and the composite of new or worsening nephropathy.
A total of 7020 patients were treated with empagliflozin (empagliflozin 10 mg: 2345, empagliflozin 25 mg: 2342, placebo: 2333) and followed for a median of 3.1 years.
The population was 72.4% Caucasian, 21.6% Asian, and 5.1% Black. The mean age was 63 years and 71.5% were male. At baseline, approximately 81% of patients were being treated with renin angiotensin system inhibitors, 65% with beta-blockers, 43% with diuretics, 89% with anticoagulants, and 81% with lipid lowering medication. Approximately 74% of patients were being treated with metformin at baseline, 48% with insulin and 43% with sulphonylurea.
About half of the patients (52.2%) had an eGFR of 60-90 ml/min/1.73 m2, 17.8% of 45-60 ml/min/1.73 m2 and 7.7% of 30-45 ml/min/1.73 m2. Mean systolic BP was 136 mmHg, diastolic BP 76 mmHg, LDL 86 mg/dL, HDL 44 mg/dL, and urinary albumin to creatinine ratio (UACR) 175 mg/g at baseline.
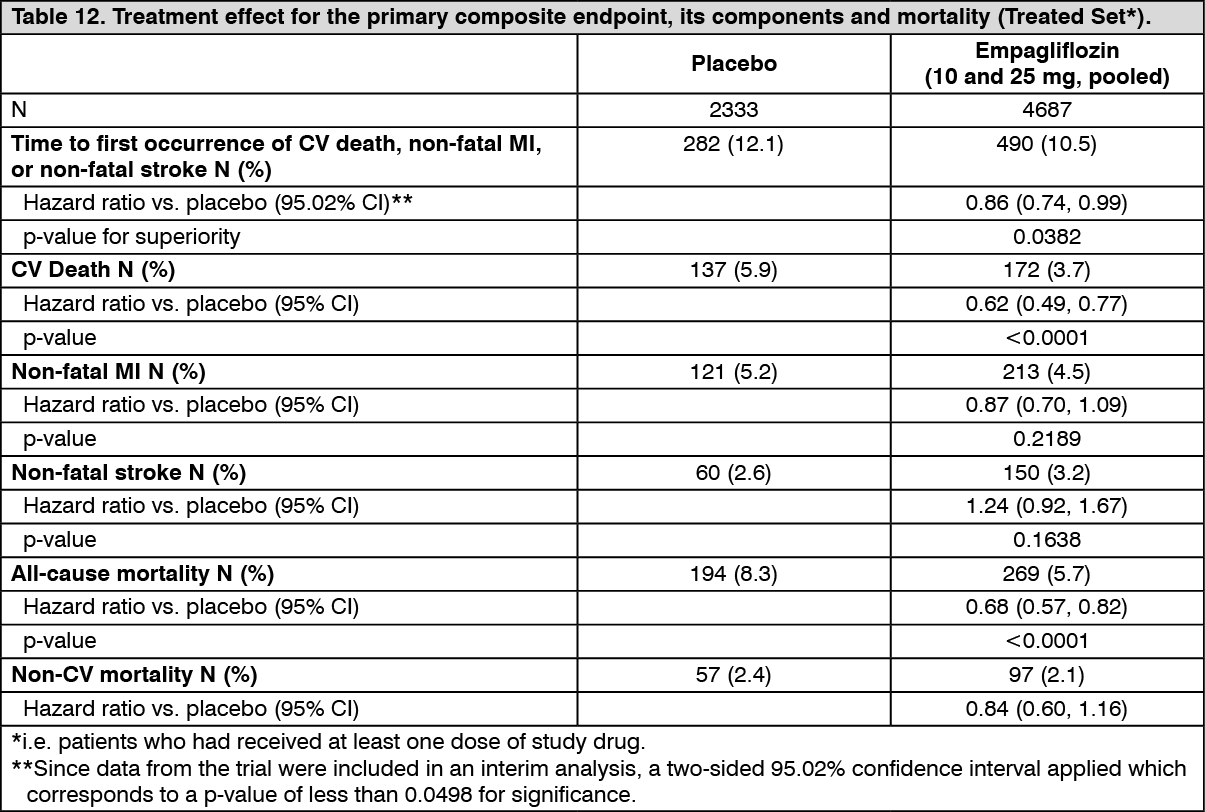
Reductions in risk of CV death and overall mortality: Empagliflozin is superior in reducing the primary composite endpoint of cardiovascular death, non-fatal MI, or non-fatal stroke compared to placebo. The treatment effect reflected a reduction in cardiovascular death with no significant change in non-fatal MI, or non-fatal stroke (Table 12 and Figure 1).
Empagliflozin also improved overall survival (Table 12 and Figure 2), which was driven by a reduction in cardiovascular death with empagliflozin. There was no statistically significant difference between empagliflozin and placebo in non-cardiovascular mortality. (See Table 12 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image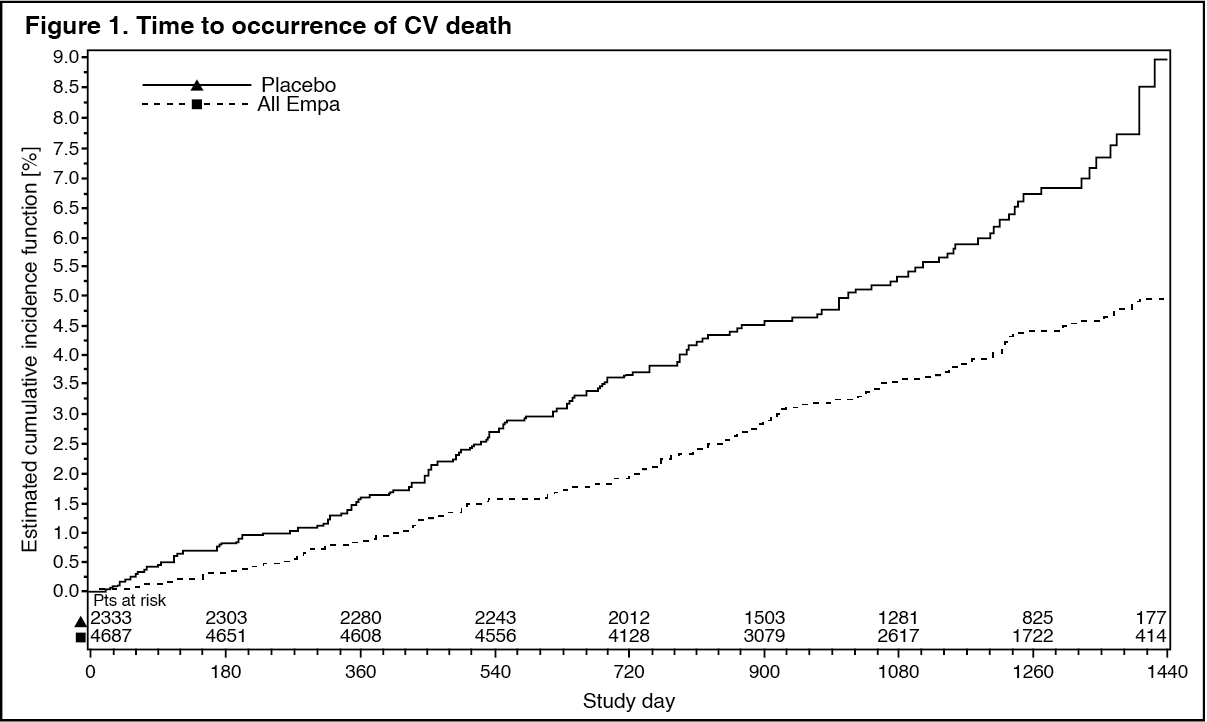 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image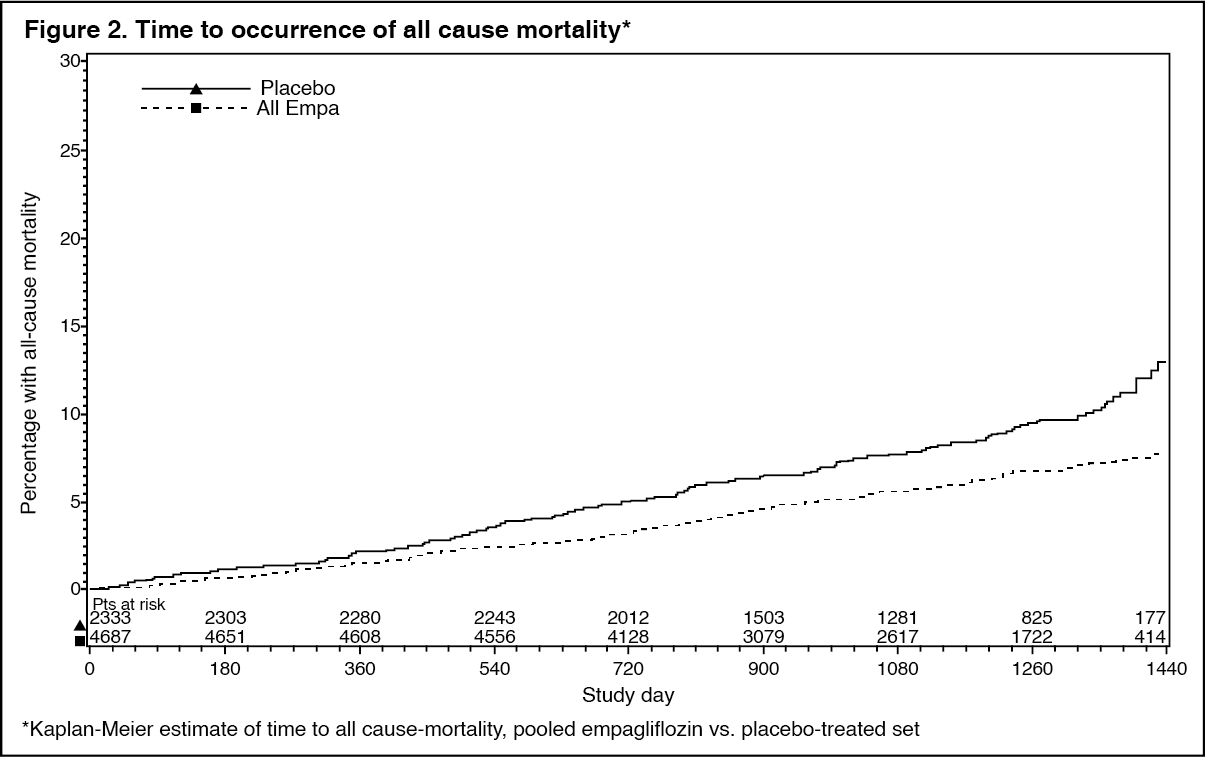 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageReductions in risk of heart failure requiring hospitalization or CV death: Empagliflozin is superior in reducing the risk of hospitalization for heart failure and cardiovascular death or hospitalization for heart failure compared with placebo (Table 13 and Figure 3). (See Table 13 and Figure 3.)
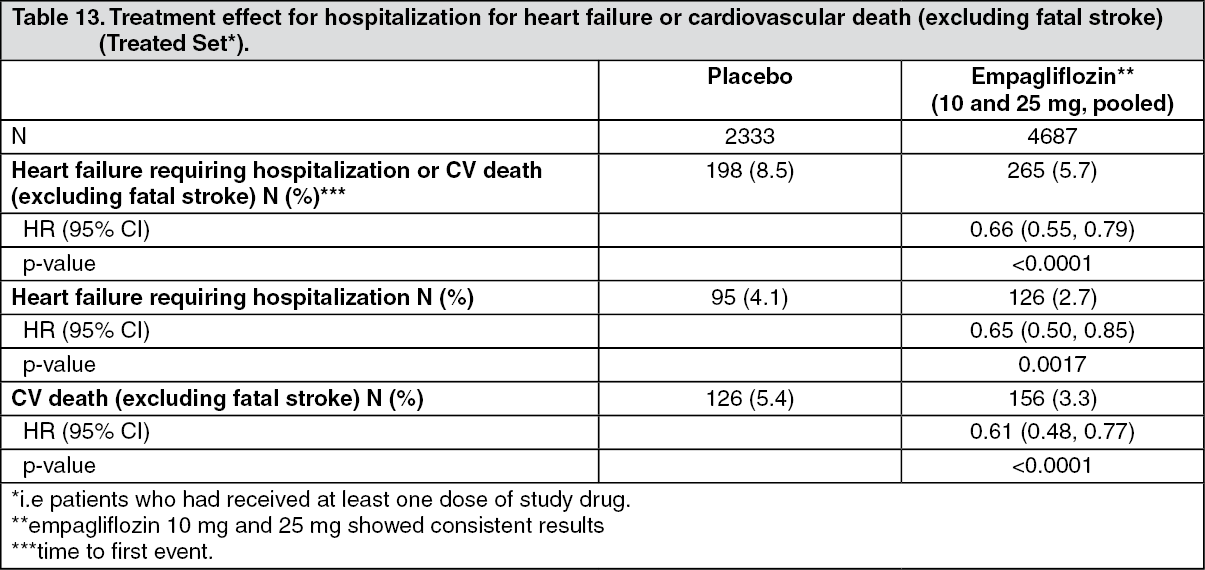 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image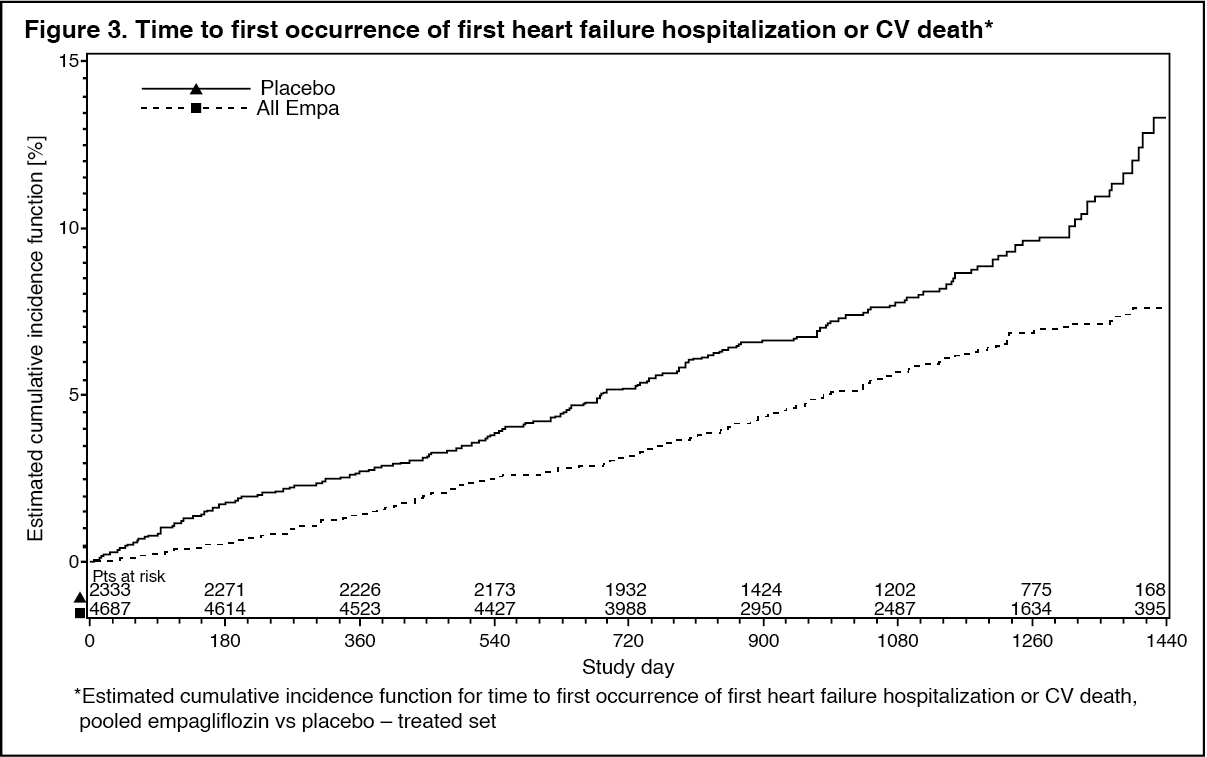 Click on icon to see table/diagram/image
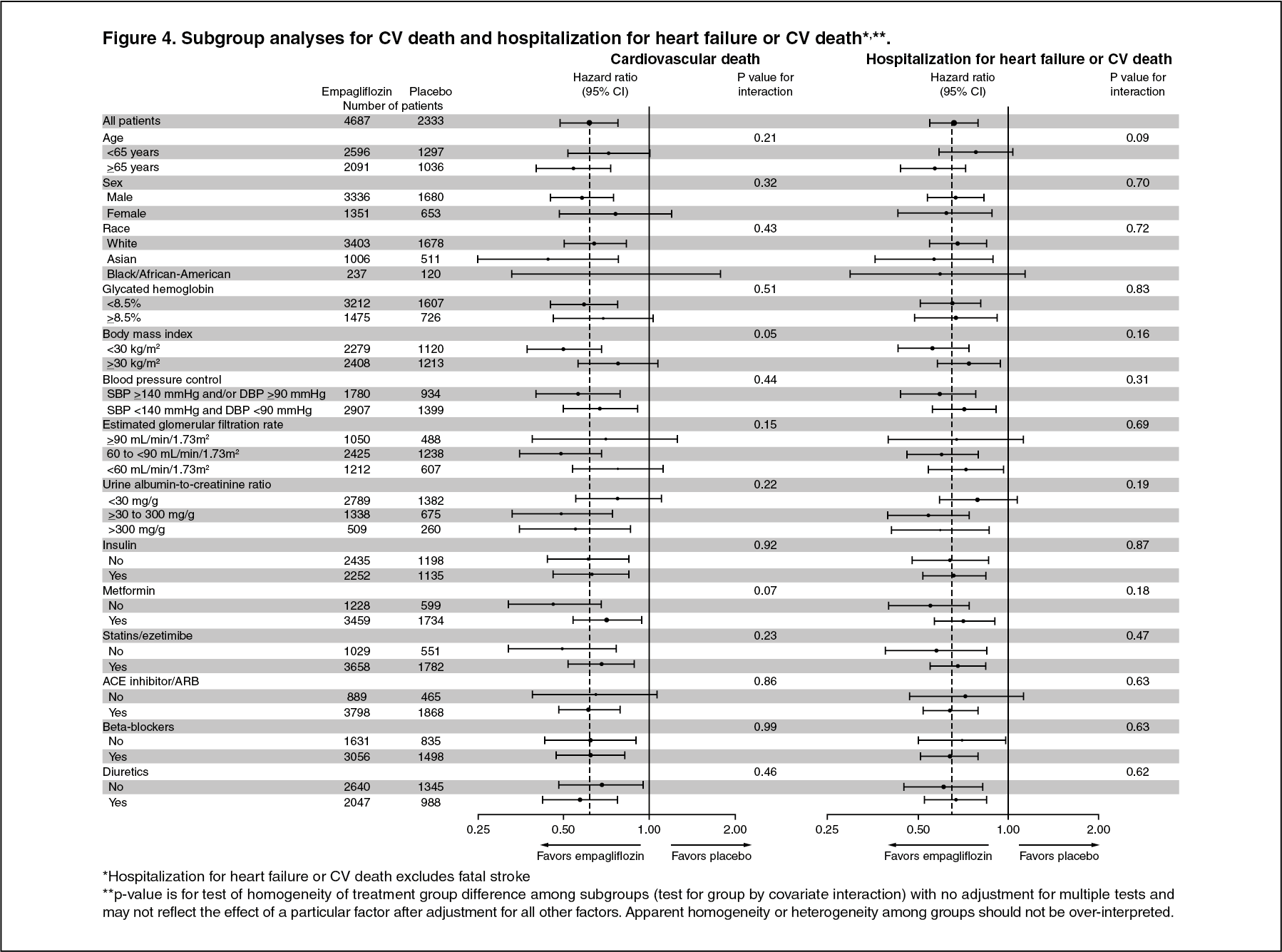
Click on icon to see table/diagram/imageThe cardiovascular benefits of empagliflozin observed were consistent across the subgroups depicted in Figure 4. (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the subgroup of patients who were on metformin at baseline, the effects on CV outcomes were consistent with the results observed in the entire study population of EMPA REG OUTCOME.
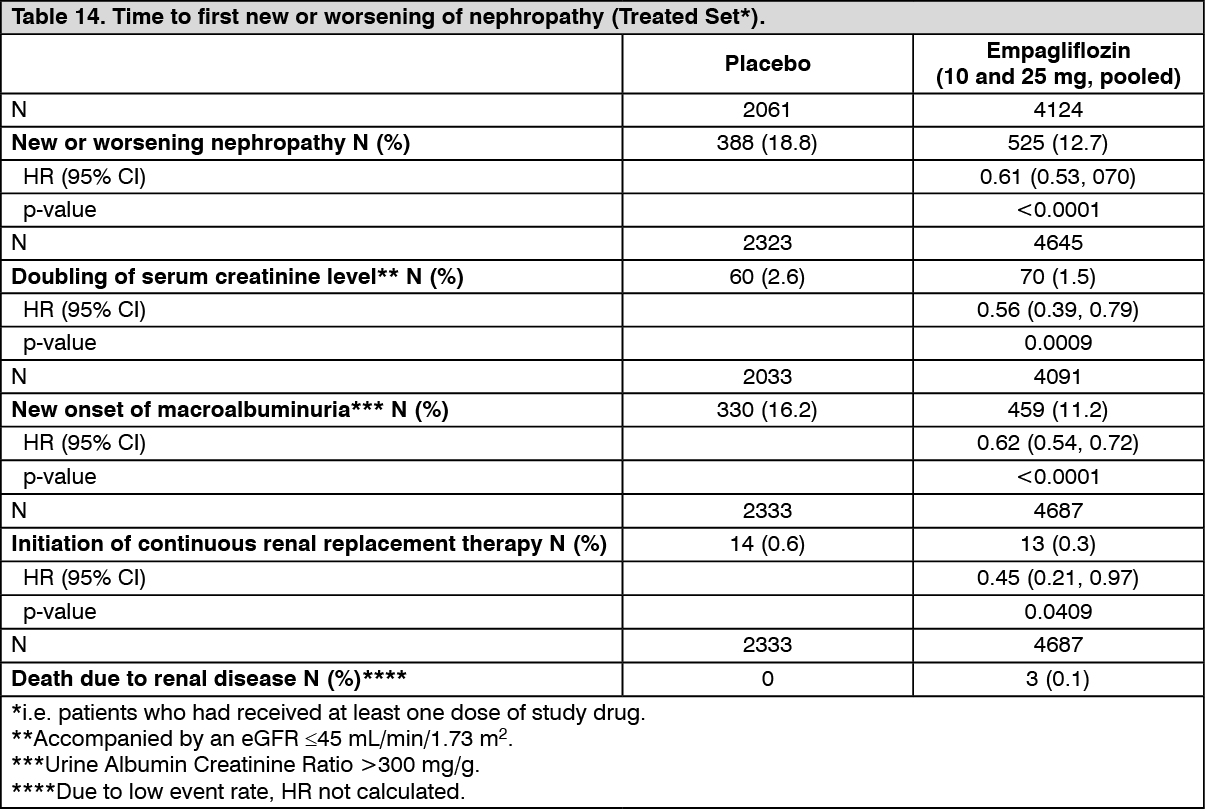
Diabetic kidney disease: In the EMPA-REG OUTCOME study population, the risk of new or worsening nephropathy (defined as onset of macroalbuminuria, doubling of serum creatinine, and initiation of renal replacement therapy (i.e. hemodialysis)) was significantly reduced in empagliflozin group compared to placebo (Table 14 and Figure 5).
Empagliflozin compared with placebo showed a significantly higher occurrence of sustained normo- or microalbuminuria in patients with baseline macroalbuminuria (HR 1.82, 95% CI 1.40, 2.37). (See Table 14 and Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image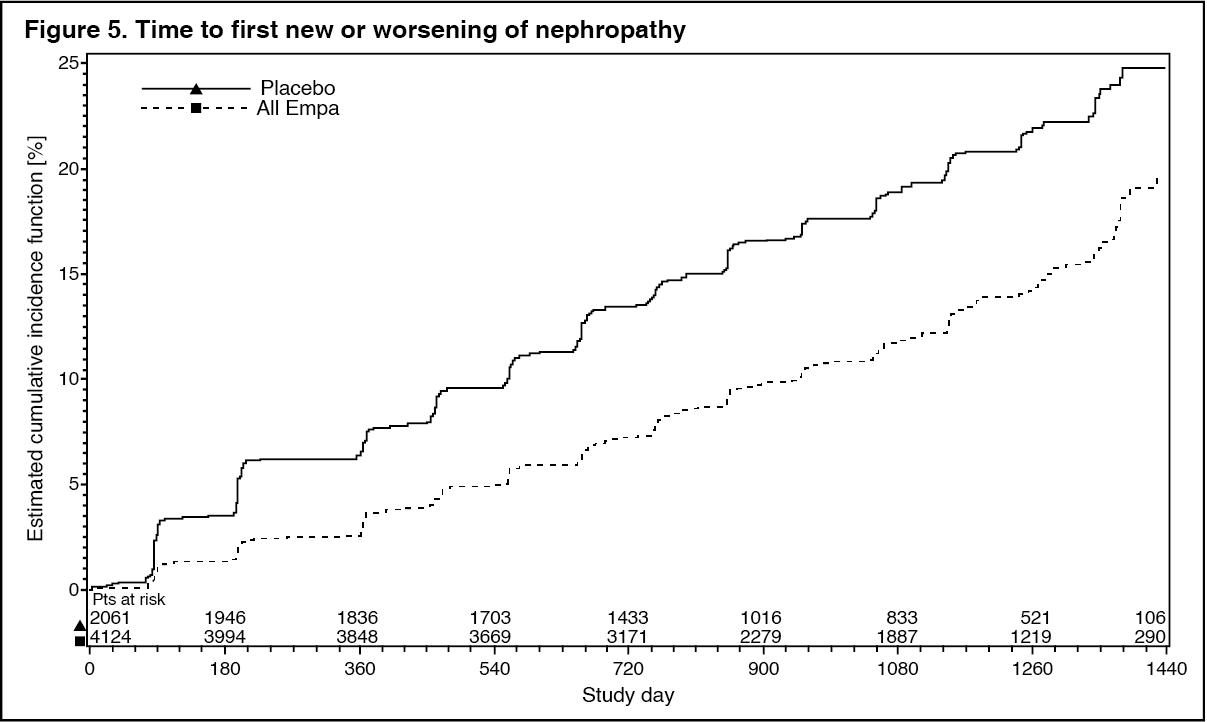 Click on icon to see table/diagram/image
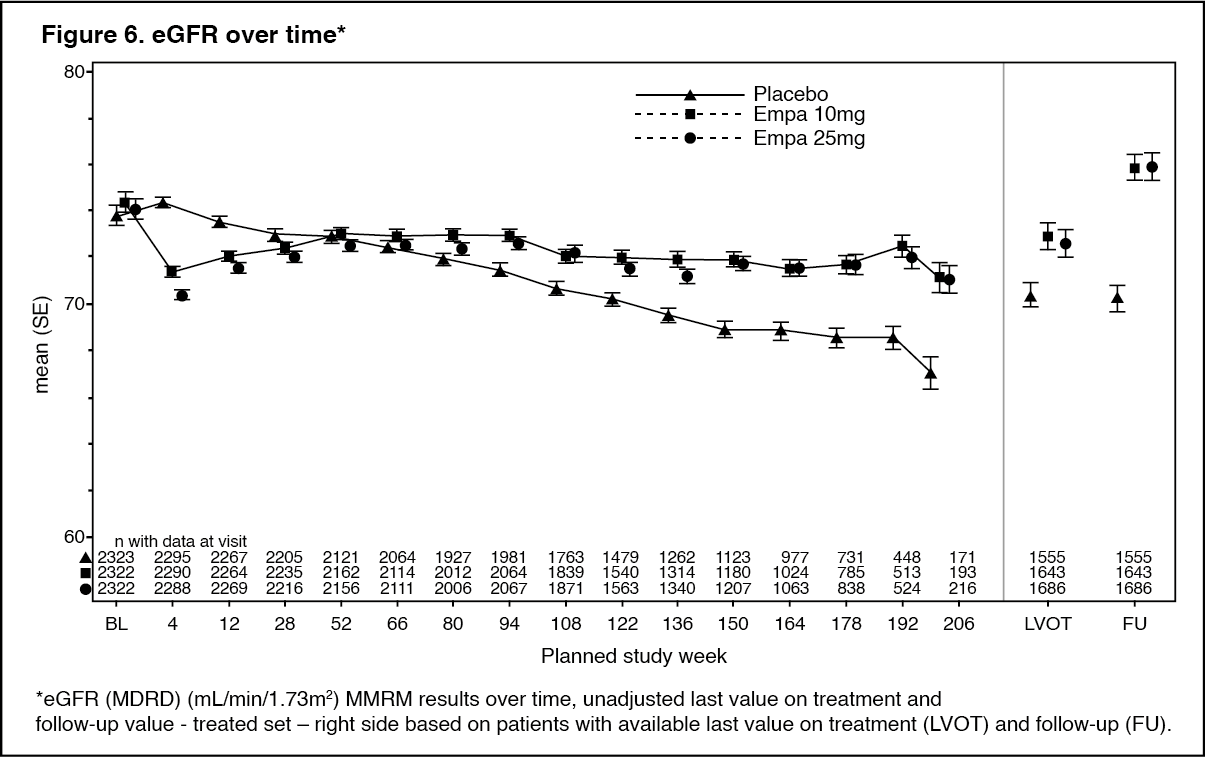
Click on icon to see table/diagram/imageTreatment with empagliflozin preserved eGFR and eGFR increased during the post treatment 4-week follow up. However, the placebo group showed a gradual decline in GFR during the course of the study with no further change during 4-week follow up. (See Figure 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the subgroup of patients who were on metformin at baseline, the effects on these renal outcomes were consistent with the results observed in the entire study population of EMPA REG OUTCOME.
Thorough QTc study: In a randomized, placebo-controlled, active-comparator, crossover study of 30 healthy subjects no increase in QTc was observed with either 25 mg or 200 mg empagliflozin.
Pharmacokinetics: JARDIANCE DUO: The results of bioequivalence studies in healthy subjects demonstrated that JARDIANCE DUO (empagliflozin/metformin hydrochloride) 5 mg/1000 mg and 12.5 mg/1000 mg combination tablets are bioequivalent to co-administration of corresponding doses of empagliflozin and metformin as individual tablets.
Administration of 12.5 mg empagliflozin/1000 mg metformin under fed conditions resulted in a 9% decrease in AUC and a 28% decrease in Cmax for empagliflozin, when compared to fasted conditions. For metformin, AUC decreased by 12% and Cmax decreased by 26% compared to fasting conditions. The observed effect of food on empagliflozin and metformin is not considered to be clinically relevant. However, as metformin is recommended to be given with meals, JARDIANCE DUO is also proposed to be given with food.
The following data are findings in studies performed with empagliflozin or metformin individually.
Empagliflozin: Absorption: The pharmacokinetics of empagliflozin have been extensively characterized in healthy volunteers and patients with T2DM. After oral administration, empagliflozin was rapidly absorbed with peak plasma concentrations occurring at a median tmax 1.5 h post-dose. Thereafter, plasma concentrations declined in a biphasic manner with a rapid distribution phase and a relatively slow terminal phase. The steady-state mean plasma AUC and Cmax were 1870 nmoL.h and 259 nmoL/L with empagliflozin 10 mg and 4740 nmoL.h/L and 687 nmoL/L with empagliflozin 25 mg once daily, respectively. Systemic exposure of empagliflozin increased in a dose-proportional manner. The single-dose and steady-state pharmacokinetics parameters of empagliflozin were similar suggesting linear pharmacokinetics with respect to time. There were no clinically relevant differences in empagliflozin pharmacokinetics between healthy volunteers and patients with type 2 diabetes mellitus.
The pharmacokinetics of 5 mg empagliflozin twice daily and 10 mg empagliflozin once daily were compared in healthy subjects. Overall exposure (AUCss) of empagliflozin over a 24-hour period with 5 mg administered twice daily was similar to 10 mg administered once daily. As expected, empagliflozin 5 mg administered twice daily compared with 10 mg empagliflozin once daily resulted in lower Cmax and higher trough plasma empagliflozin concentrations (Cmin).
Administration of 25 mg empagliflozin after intake of a high-fat and high calorie meal resulted in slightly lower exposure; AUC decreased by approximately 16% and Cmax decreased by approximately 37%, compared to fasted condition. The observed effect of food on empagliflozin pharmacokinetics was not considered clinically relevant and empagliflozin may be administered with or without food.
Distribution: The apparent steady-state volume of distribution was estimated to be 73.8 L, based on a population pharmacokinetic analysis. Following administration of an oral [14C]-empagliflozin solution to healthy subjects, the red blood cell partitioning was approximately 36.8% and plasma protein binding was 86.2%.
Biotransformation: No major metabolites of empagliflozin were detected in human plasma and the most abundant metabolites were three glucuronide conjugates (2-O-, 3-O-, and 6-O-glucuronide). Systemic exposure of each metabolite was less than 10% of total drug-related material. In vitro studies suggested that the primary route of metabolism of empagliflozin in humans is glucuronidation by the uridine 5'-diphospho-glucuronosyltransferases, UGT1A3, UGT1A8, UGT1A9, and UGT2B7.
Elimination: The apparent terminal elimination half-life of empagliflozin was estimated to be 12.4 h and apparent oral clearance was 10.6 L/h based on the population pharmacokinetic analysis. The inter-subject and residual variabilities for empagliflozin oral clearance were 39.1% and 35.8%, respectively. With once-daily dosing, steady-state plasma concentrations of empagliflozin were reached by the fifth dose. Consistent with the half-life, up to 22% accumulation, with respect to plasma AUC, was observed at steady-state. Following administration of an oral [14C]-empagliflozin solution to healthy subjects, approximately 95.6% of the drug related radioactivity was eliminated in faeces (41.2%) or urine (54.4%). The majority of drug related radioactivity recovered in faeces was unchanged parent drug and approximately half of drug related radioactivity excreted in urine was unchanged parent drug.
Specific Populations: Renal Impairment: In patients with mild (eGFR: 60 - < 90 mL/min/1.73 m2), moderate (eGFR: 30 - < 60 mL/min/1.73 m2), severe (eGFR: < 30 mL/min/1.73 m2) renal impairment and patients with kidney failure/ESRD patients, AUC of empagliflozin increased by approximately 18%, 20%, 66%, and 48%, respectively, compared to subjects with normal renal function. Peak plasma levels of empagliflozin were similar in subjects with moderate renal impairment and kidney failure/ESRD compared to patients with normal renal function.
Peak plasma levels of empagliflozin were roughly 20% higher in subjects with mild and severe renal impairment as compared to subjects with normal renal function. In line with the Phase I study, the population pharmacokinetic analysis showed that the apparent oral clearance of empagliflozin decreased with a decrease in eGFR leading to an increase in drug exposure. Based on pharmacokinetics, no dosage adjustment is recommended in patients with renal insufficiency.
Hepatic Impairment: In subjects with mild, moderate, and severe hepatic impairment according to the Child-Pugh classification, AUC of empagliflozin increased approximately by 23%, 47%, and 75% and Cmax by approximately 4%, 23%, and 48%, respectively, compared to subjects with normal hepatic function. Based on pharmacokinetics, no dosage adjustment is recommended in patients with hepatic impairment.
Body Mass Index (BMI): No dosage adjustment is necessary based on BMI. Body mass index had no clinically relevant effect on the pharmacokinetics of empagliflozin based on the population pharmacokinetic analysis.
Gender: No dosage adjustment is necessary based on gender. Gender had no clinically relevant effect on the pharmacokinetics of empagliflozin based on the population pharmacokinetic analysis.
Race: No dosage adjustment is necessary based on race. Based on the population pharmacokinetic analysis, AUC was estimated to be 13.5% higher in Asian patients with a BMI of 25 kg/m2 compared to non-Asian patients with a BMI of 25 kg/m2.
Geriatric: Age did not have a clinically meaningful impact on the pharmacokinetics of empagliflozin based on the population pharmacokinetic analysis.
Paediatric: Studies characterizing the pharmacokinetics of empagliflozin in paediatric patients have not been performed.
Metformin: Absorption: After an oral dose of metformin, Tmax is reached in 2.5 hours. Absolute bioavailability of a 500 mg or 850 mg metformin hydrochloride tablet is approximately 50-60% in healthy subjects. After an oral dose, the non-absorbed fraction recovered in faeces was 20-30%.
After oral administration, metformin hydrochloride absorption is saturable and incomplete. It is assumed that the pharmacokinetics of metformin hydrochloride absorption are non-linear.
At the recommended metformin hydrochloride doses and dosing schedules, steady-state plasma concentrations are reached within 24 to 48 hours and are generally less than 1 μg/mL. In controlled clinical trials, maximum metformin hydrochloride plasma levels (Cmax) did not exceed 5 μg/mL, even at maximum doses.
Food decreases the extent and slightly delays the absorption of metformin hydrochloride. Following administration of a dose of 850 mg, a 40% lower plasma peak concentration, a 25% decrease in AUC (area under the curve) and a 35 minute prolongation of the time to peak plasma concentration were observed. The clinical relevance of these decreases is unknown.
Distribution: Plasma protein binding is negligible. Metformin hydrochloride partitions into erythrocytes. The blood peak is lower than the plasma peak and appears at approximately the same time. The red blood cells most likely represent a secondary compartment of distribution. The mean volume of distribution (Vd) ranged between 63-276 L.
Biotransformation: Metformin hydrochloride is excreted unchanged in the urine. No metabolites have been identified in humans.
Elimination: Renal clearance of metformin hydrochloride is > 400 mL/min, indicating that metformin hydrochloride is eliminated by glomerular filtration and tubular secretion. Following an oral dose, the apparent terminal elimination half-life is approximately 6.5 hours. When renal function is impaired, renal clearance is decreased in proportion to that of creatinine and thus the elimination half-life is prolonged, leading to increased levels of metformin hydrochloride in plasma.
Special populations: Renal impairment: The available data in subjects with moderate renal insufficiency are scarce and no reliable estimation of the systemic exposure to metformin in this subgroup as compared to subjects with normal renal function could be made. Therefore, the dose adaptation should be made upon clinical efficacy/tolerability considerations (see Dosage & Administration).
Paediatric: Single dose study: After single doses of metformin 500 mg, paediatric patients, have shown a similar pharmacokinetic profile to that observed in healthy adults.
Multiple dose study: After repeated doses of 500 mg twice daily for 7 days in paediatric patients the peak plasma concentration (Cmax) and systemic exposure (AUC0-t) were approximately 33% and 40% lower, respectively, compared to diabetic adults who received repeated doses of 500 mg twice daily for 14 days. As the dose is individually titrated based on glycaemic control, this is of limited clinical relevance.
TOXICOLOGY: Empagliflozin and Metformin: General toxicity studies in rats up to 13 weeks were performed with the combination of empagliflozin and metformin. In a 13 week combination study with empagliflozin and metformin in rats the No-observed-adverse-effect-level (NOAEL) was based on hypochloremia seen at exposures of approximately 24- and 9-times the clinical AUC exposure of empagliflozin associated with the 10 and 25 mg doses, respectively.
An embryofetal development study in pregnant rats did not indicate a teratogenic effect attributed to the co-administration of empagliflozin and metformin at exposures of approximately 35- and 14-times the clinical AUC exposure of empagliflozin associated with the 10 and 25 mg doses, respectively, and 4-times the clinical AUC exposure of metformin associated with the 2000 mg dose. At dose levels of 600 mg/kg/day, associated with 8-times the maximum recommended human dose (MRHD) of metformin in humans, teratogenicity of metformin was observed.
The following data are findings in studies performed with empagliflozin or metformin individually.
Empagliflozin: In general toxicity studies in rodents and dogs, signs of toxicity were observed at exposures greater than or equal to 10-times the clinical dose of 25 mg. Most toxicity was consistent with secondary pharmacology related to urinary glucose loss and included decreased body weight and body fat, increased food consumption, diarrhoea, dehydration, decreased serum glucose and increases in other serum parameters reflective of increased protein metabolism, gluconeogenesis and electrolyte imbalances, urinary changes such as polyuria and glucosuria, and microscopic changes in kidney.
Carcinogenicity: Empagliflozin did not increase the incidence of tumours in female rats at doses up to the highest dose of 700 mg/kg/day, which corresponds to approximately 72- and 182- times the clinical AUC exposure associated with the 25 mg and 10 mg doses, respectively. In male rats, treatment-related benign vascular proliferative lesions (hemangiomas) of the mesenteric lymph node were observed at 700 mg/kg/day, which corresponds to approximately 42- and 105-times the clinical exposure associated with the 25 mg and 10 mg doses, respectively. These tumours are common in rats and are unlikely to be relevant to humans. Empagliflozin did not increase the incidence of tumours in female mice at doses up to 1000 mg/kg/day, which corresponds to approximately 62- and 158-times the clinical exposure associated with the 25 mg and 10 mg doses, respectively. Empagliflozin induced renal tumours in male mice at 1000 mg/kg/day, which corresponds to approximately 45- and 113-times the clinical exposure associated with the 25 mg and 10 mg doses, respectively. The mode of action for these tumours is dependent on the natural predisposition of the male mouse to renal pathology and a metabolic pathway not reflective of humans. The male mouse renal tumours are considered not relevant to humans.
Genotoxicity: Empagliflozin is not genotoxic.
Reproduction Toxicity: Nonclinical studies show that empagliflozin crosses the placenta during late gestation to a very limited extent but do not indicate direct or indirect harmful effects with respect to early embryonic development. Empagliflozin administered during the period of organogenesis was not teratogenic at doses up to 300 mg/kg in the rat or rabbit, which corresponds to approximately 48- and 122-times or 128- and 325-times the clinical dose of empagliflozin based on AUC exposure associated with the 25 mg and 10 mg doses, respectively. Doses of empagliflozin causing maternal toxicity in the rat also caused the malformation of bent limb bones at exposures approximately 155- and 393-times the clinical dose associated with the 25 mg and 10 mg doses, respectively. Maternally toxic doses in the rabbit also caused increased embryofetal loss at doses approximately 139- and 353-times the clinical dose associated with the 25 mg and 10 mg doses, respectively.
In pre- and postnatal toxicity studies in rats, reduced weight gain in offspring was observed at maternal exposures approximately 4- and 11-times the clinical dose associated with the 25 mg and 10 mg doses, respectively.
In a juvenile toxicity study in the rat, when empagliflozin was administered from postnatal day 21 until postnatal day 90, non-adverse, minimal to mild renal tubular and pelvic dilation in juvenile rats was seen only at 100 mg/kg/day, which approximates 11-times the maximum clinical dose of 25 mg. These findings were absent after a 13 weeks drug-free recovery period.
Metformin: Nonclinical data reveal no special hazard for humans based on conventional studies on safety pharmacology, genotoxicity, and carcinogenic potential. In a 2 week metformin only study and 2 and 13-week toxicity empagliflozin/metformin studies in rats, metformin related toxicity was seen in heart, liver, kidneys, salivary glands, ovaries, gastrointestinal tract and adrenal glands at dosages associated with a systemic exposure of 5 times the MRHD or higher.
Metformin was not teratogenic in rats at a dose of 200 mg/kg/day associated with a systemic exposure of 4 times the MRHD (2000 mg metformin). At higher doses (500 and 1000 mg/kg/day, associated with 11 and 23 times the MRHD), teratogenicity of metformin was observed in the rat which was mostly evident as an increase in the number of skeletal malformations.